Im Zuge der Umstellung auf die MDR (Medical Device Regulation) wurde auch die Normenreihe der DIN EN ISO 10993 aktualisiert, insbesondere die DIN EN ISO 10993-1. In der MDR wird der Nachweis auf Biokompatibilität von Materialien gefordert, die mit dem Patienten oder Anwender in Kontakt kommen. Die entsprechenden Anforderungen werden in der ISO 10993 näher aufgeschlüsselt.
In dieser wird Biokompatibilität wie folgt definiert: „Fähigkeit eines Medizinprodukts oder Materials, mit einer angemessenen Host-Reaktion Leistung in einer spezifischen Anwendung zu erbringen“
Im Rahmen des biologischen Risikomanagementprozesses zur Evaluierung der Biokompatibilität stehen nun Literaturrecherche, physikalisch-chemische Analysen sowie in-vitro Testungen im Vordergrund, sodass Tierversuche zur Bewertung der Biokompatibilität weitestgehend reduziert werden können.
Biokompatible Materialien können des Weitern eingeteilt werden in:
• Biotolerant: es gibt kaum oder nur geringfügige, unerwünschte Nebenreaktionen
• Bioinert: idealerweise keine Wechselwirkungen zwischen Fremdkörper und Organismus; in der Realität wird derartiges Material ohne Abstoßungsreaktion vom Körper eingekapselt
• Bioaktiv: es gibt erwünschte Wechselwirkungen, die durch bestimmte Strukturen oder biologische Signale gelenkt werden;
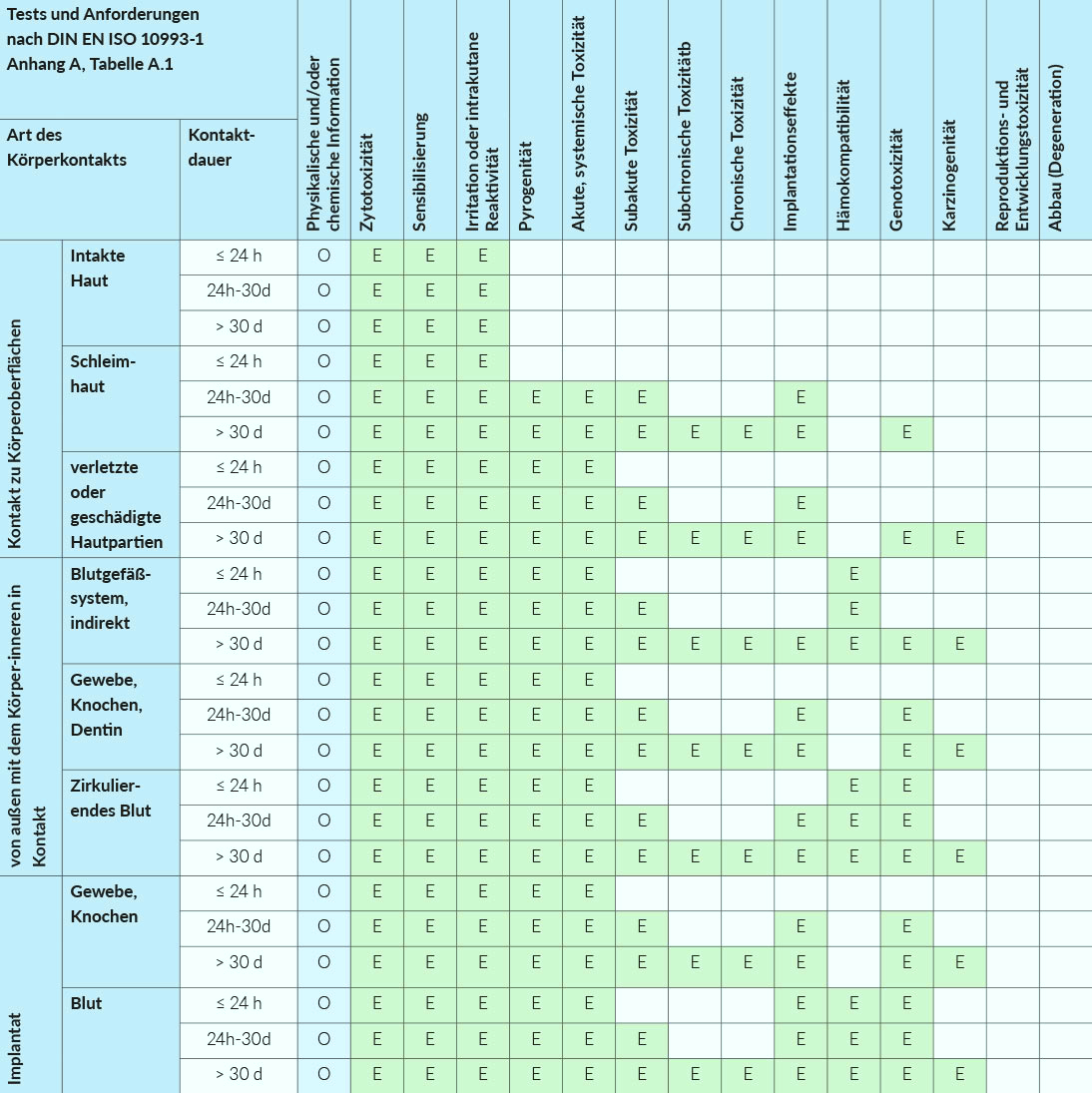
Je nach Kategorie und Art des Einsatzes ändert sich der Prüfumfang und durch die Versuchsergebnisse wird dann das Gefährdungspotential bewertet.
In der nachfolgenden Grafik steht:
O für obligatorisch
(der Hersteller und Inverkehrbringer muss die Bestandteile des Medizinproduktes kennen)
E für empfohlen
(der Hersteller und Inverkehrbringer muss mindestens eine toxikologische Endpunktbestimmung der Substanzen vornehmen)